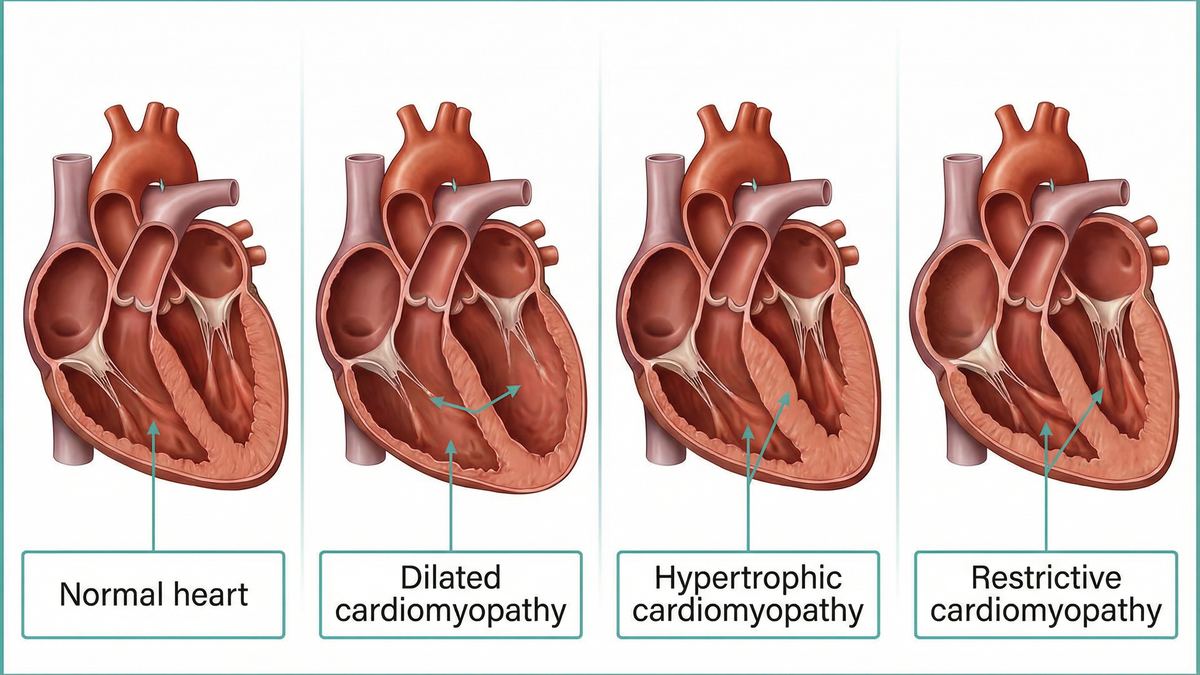
| Hypertrophic (HCM) |
The heart muscle (especially the septum) becomes abnormally thick. |
Often genetic. Can cause obstruction of blood flow out of the heart (obstructive HCM). A leading cause of sudden cardiac death in young athletes. |
1 in 200 to 1 in 500 people [1]. |
| Dilated (DCM) |
The left ventricle becomes enlarged (dilated) and the walls become thin. |
The heart muscle weakens and cannot pump effectively (reduced ejection fraction). The most common reason for heart transplantation. |
Approximately 1 in 2,500 people [1]. |
| Restrictive (RCM) |
The heart muscle becomes rigid and stiff, though not necessarily thickened. |
The heart cannot relax and fill with blood properly during diastole. Often caused by infiltrative diseases like cardiac amyloidosis. |
Rare; accounts for 2% to 5% of all cardiomyopathies [1]. |
| Arrhythmogenic (ARVC) |
Heart muscle tissue in the right ventricle is replaced by fat and scar tissue. |
Highly arrhythmogenic, increasing the risk of dangerous irregular heartbeats. Strongly linked to genetic mutations in desmosomal genes. |
Rare; primarily affects young adults and athletes. |
| Left Ventricular Non-Compaction (LVNC) |
The lower left chamber has a spongy, "trabeculated" appearance. |
A rare congenital condition where the heart muscle does not develop normally in utero. Can lead to heart failure and blood clots. |
Rare congenital anomaly. |